药物临床试验数据递交指导原则(试行)
一、背景与目的
药物临床试验数据是申办方向监管机构递交的重要资料之一,对于监管机构和申办方来说都是宝贵的资源。规范地收集、整理、分析和呈现临床试验数据对于提高药物临床研发的效率和质量、缩短审评时间具有重要的作用,并且有利于药品全生命周期管理,促进研发或监管的信息互通或共享。
申办方递交的临床试验数据若不遵循一定的规范,熟悉和理解数据结构及内容将占用大量的审评资源。某些情况下,申办方或监管机构可能需要针对多来源的临床试验数据进行汇总分析,如果数据没有规范化,整合利用也几乎难以实现。
临床试验数据相关的申报资料通常包括数据库及其相应的数据说明文件、数据审阅说明、程序代码和注释病例报告表(annotated Case Report Form,aCRF)。本指导原则主要对临床试验数据递交的内容及格式提出具体要求,旨在指导申办方规范递交临床试验数据及相关资料,同时有助于数据管理、统计分析等相关从业人员更好地开展临床试验中的相关工作。
本指导原则主要适用于以支持药品注册上市为目的的关键性临床试验,也可供以非注册为目的的临床试验参考使用。本指导原则基于国际监管机构数据递交要求以及国内现状制定,申办方应基于本指导原则要求准备相关资料。鼓励申办方参照临床数据交换标准协会(Clinical Data Interchange Standards Consortium,CDISC)标准递交临床试验数据及相关的申报资料。随着临床试验数据标准的发展以及对其认识与实践的提高,本指导原则会酌情修订完善。
二、临床试验数据相关资料及其说明
(一)原始数据库
原始数据库通常包含从病例报告表和外部文件中直接收集的原始数据,还可能包含极少量的衍生数据,如序号。原始数据库中的缺失数据不应进行填补。为满足数据递交的要求,直接收集的数据可能需要进行必要的标准化或编码,例如调整数据库中数据集名称/标签/结构、数据集中变量名称/标签,或在适用的情况下对变量值进行标准化编码,如监管活动医学词典(Medical Dictionary for Regulatory Activities,MedDRA)等。如果申办方参照CDISC标准递交数据,则可将原始数据标准模型(Study Data Tabulation Model,SDTM)数据库视为原始数据库。
原始数据库通常包含多个原始数据集,原始数据集应按主题进行组织并命名,数据集通常以两个英文字母组成的代码命名,如人口学(dm)、不良事件(ae)、实验室检查(lb)等数据集。临床试验中常见的原始数据集命名详见附录1。
所有递交的原始数据集必须包含研究标识符(STUDYID)变量;反映各受试者观测结果的数据集(如附录1中的dm、ae、lb等数据集)中还必须包含受试者唯一标识符(USUBJID)变量;另外,受试者标识符(SUBJID)变量必须包含在dm数据集中。常用到的标识符举例说明如下:
研究标识符:变量名为STUDYID,字符型,研究的唯一标识符,即研究编号。
受试者唯一标识符:变量名为USUBJID,字符型,每一受试者在同一产品的整个试验申请(包含多个临床研究)过程中应当赋予相同的唯一标识符。在所有数据集(包括原始数据集与分析数据集)中,同一个受试者应当有完全相同的唯一标识符。当受试者参加了多个研究,各个研究之间的USUBJID应当保持一致。遵循这一规则对于合并同一受试者在不同研究中的数据尤其重要(如随机对照试验或扩展性研究)。
受试者标识符:变量名为SUBJID,字符型,SUBJID是参加试验的受试者的标识符。
访视名称(VISIT,字符型)和访视编号(VISITNUM,数值型)等时间变量应包含在适用的数据集中。计划访视的VISITNUM应根据时间顺序从小到大赋值,并与VISIT一一对应。
(二)分析数据库
分析数据库是为统计分析衍生新建的数据库,用于产生和支持临床总结报告等文件中的统计分析结果。分析数据库中一般包含原始数据及基于原始数据按照一定规则衍生的数据,如对缺失数据填补后的数据等。如果申办方参照CDISC标准递交数据,则可将分析数据标准模型(Analysis Data Model,ADaM)数据库视为分析数据库。
分析数据库通常包含多个分析数据集。构建分析数据集时,可能会将收集和衍生的数据(来自各原始数据集或其它分析数据集)合并到一个数据集中,构建时应遵循以下原则:①用于支持统计分析的分析数据集,其内容与来源必须清晰。②分析数据集必须具备可追溯性,数据衍生的具体规则应在相应的数据说明文件中加以详细说明。③分析数据集结构和内容应满足只需要很少的编程即可进行统计分析。
分析数据库应包含分析时所需的所有变量,包括衍生变量,且所有衍生变量均应能通过原始数据库及其它支持性数据文件生成。分析数据集通常以“adxxxxxx”命名,分析数据集的命名应尽量与原始数据集保持对应,如:adcm、adae、adlb等。
受试者水平分析数据集(命名为adsl)是必不可少的一个分析数据集。在该数据集中,每一受试者应仅有一条记录,内容应包括但不限于人口学、重要的基线特征/分层因素、治疗组、预后因素、重要日期、分析人群划分等信息。
对于有些终点(如某些量表评分),从原始数据集到可用于最终统计分析的分析数据集需要经过一系列衍生过程,为方便最终分析数据集创建而衍生的中间变量/数据集必要时也应一同包含在分析数据库中。
(三)数据说明文件
递交的原始数据库和分析数据库必须有相应的数据说明文件。数据说明文件是一份用来描述递交数据的文件,至少应包含递交数据库中各数据集名称、标签、基本结构描述及每一数据集中各变量的名称、标签、类型、来源或衍生过程。
数据说明文件是监管机构审评时准确理解递交数据内容最重要的文件之一。申办方应确保每个变量的编码列表和来源都有清晰的定义,并且易于查找。如果使用外部词典,需要在数据说明文件中指明所用的词典及版本。需要通过数据说明文件建立起数据间良好的可追溯性(如:原始数据集与CRF、分析数据集与原始数据集之间),以便于监管机构的审阅。申办方需要在数据说明文件中提供相关细节,尤其是和衍生变量相关的详细说明,必要时可使用关键程序代码辅助说明。
数据说明文件一般为可扩展标记语言(Extensible Mark-up Language,XML)或便携文档格式(Portable Document Format,PDF)文件。如递交XML格式数据说明文件,对应的可扩展样式表语言(Extensible Stylesheet Language,XSL)文件也应一并递交。
(四)数据审阅说明
为了帮助审评人员更好地理解与使用递交的数据,鼓励申办方递交数据审阅说明。数据审阅说明是对数据说明文件的进一步补充,其内容包括但不限于研究数据使用说明、临床总结报告与数据之间的关系、研究文档(如试验方案、统计分析计划、临床总结报告等)中部分关键信息、所递交程序代码的使用说明、数据集所用编码(如utf-8、euc-cn等)及其它特殊情形说明等。数据审阅说明并不旨在取代数据库的数据说明文件,而是通过文档描述的方式来帮助审评人员更准确、高效的理解与使用所递交的数据库、相关术语、程序代码及数据说明文件信息等。数据审阅说明应采用PDF文件。
(五)注释病例报告表
注释病例报告表是在空白CRF的基础上,对采集的受试者数据(电子化的或者纸质的)信息单元(即字段信息)与递交原始数据集中对应的变量或变量值之间映射关系的具体描述。aCRF文件应采用PDF文件。
实践中,CRF中可能会收集一些递交数据库中没有的数据内容,这类数据应在aCRF上明确标注为“不递交”(“NOT SUBMITTED”),并在数据审阅说明中阐明不递交这些数据的理由。
(六)程序代码
申办方需要递交的程序代码包括但不限于:分析数据集中衍生变量的衍生过程、疗效指标分析结果的生成过程等。申报资料中递交的程序代码应当易懂、可读性强,建议提供充分的注释、避免外部(宏)程序调用。程序代码一般采用TXT文件。
三、临床试验数据相关资料的格式
(一)便携文档格式
便携文档格式(PDF)是一种开放文档格式,其独立于应用软件、硬件和操作系统。申报递交资料中遵循国际人用药品注册技术要求协调会(International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use,ICH)电子通用技术文档(Electronic Common Technical Document,eCTD)格式要求的其它文档可采用PDF文件。建议使用PDF 1.4以上的版本进行文档的递交。所有PDF文件都应以.pdf作为文件扩展名。
(二)可扩展标记语言格式
可扩展标记语言(XML)是由国际万维网协会(World Wide Web Consortium,W3C)定义的一种数据交换语言。它可以被任何文本编辑器打开、编辑和创建,用来传输和存储数据。XML格式文件能够便捷地在不同系统之间进行信息交互。所有XML格式文件必需以.xml作为文件扩展名。
(三)纯文本格式
纯文本格式文档(TXT)具有格式简单、体积小、存储简单方便等诸多特点,也是计算机及许多移动终端支持的通用文件格式。所有TXT文件都应以.txt作为文件扩展名。
(四)研究数据传输格式
申报资料中的数据集通常采用SAS数据传输格式(SAS Transport Format,简称XPT)。一个XPT文件对应一个数据集,数据集名称需要与XPT文件名保持一致,其文件后缀名统一为.xpt。例如不良事件数据集ae.xpt,既往与合并用药数据集cm.xpt等。建议采用XPT第5版本(简称XPT V5)或以上版本作为数据递交格式。申办方应说明所用编码(如utf-8、euc-cn等),以避免所递交的数据集出现乱码的情形。
(五)数据集拆分
当数据库中单个数据集因存储大小不满足申报资料相关要求而需要拆分时,可仅递交拆分后的数据集。在数据审阅说明中,应详细说明数据集的拆分规则及合并的详细步骤,以确保审评人员能够生成与申办方拆分前相同的数据集。
(六)数据集名称、变量名称及变量长度
对数据集名称和变量名称要求如下:
数据集名称只能包含小写英文字母和数字,并且必须以小写字母开头。且数据集名称的最大长度为8个字节。
变量名称只能包含大写英文字母、下划线和数字,并且必须以字母开头。且变量名称的最大长度为8个字节。
每个字符型变量的长度,应该设置为在此研究所有数据集里该变量的最大实际变量值长度,有效控制文件的大小。
(七)数据集标签及变量标签
为了便于审阅,数据集标签和变量标签应使用中文,建议长度不超过40字节,必要时可以包含英文字符、下划线或数字,但不能以数字开头,另外,也不能包含下列情形:
- 不成对的 半角或全角 单引号、双引号
- 不成对的 半角或全角 括号
- 特殊字符(如 ‘>’、‘<’)
四、其它相关事项
(一)试验数据的可追溯性
审评中的一个重要环节是对数据来源的准确理解,即数据的可追溯性。可追溯性为审评人员理解统计分析结果(临床总结报告中的报表)、分析数据及与原始数据之间的关系提供了技术许可。
数据的可追溯性确保审评人员能够准确地:
- 理解分析数据集的构 建
- 确定用于衍生变量的观测记录以及相应算法
- 理解相关统计结果的计算方法
- 建立从原始数据到相应报表之间的关联
申办方在递交数据库时应确保监管部门能够利用原始数据库衍生出与申办方一致的分析数据库,利用分析数据库能够直接重现出与申办方一致的统计分析结果。可追溯性还可以通过提供数据从收集阶段到递交阶段的详细流程图来辅助解释。
(二)电子通用技术文档下的数据文件
在采用eCTD申报时,所有文档、试验数据和相关支持性文件需要按照指定的文件夹结构进行整理。所有递交的文件都应该在正确的文件夹内,并使用适当的研究标签文件(Study Tagging File,STF)进行标识。STF和文件夹结构见附录2和附录3。
(三)外文数据库
临床试验数据相关的申报资料应以中文为主,申报资料不同文件之间的中文表述应保持一致,例如分析数据集中的不良事件名称与临床总结报告报表中的不良事件名称应互相对应。为了提高审阅效率,临床试验数据相关的申报资料由外文翻译为中文的最低要求如下:
递交数据库中至少以下内容应为中文:数据集标签和变量标签;在临床总结报告等文件中出现的不良事件名称、合并用药名称、病史名称。
数据说明文件中至少以下内容应为中文:数据库中各数据集的描述/标签和说明;数据集中各变量的描述/标签和衍生过程;涉及疗效指标的取值或编码列表。
注释病例报告表中至少以下内容应为中文:为了收集数据所设计的问题描述;涉及疗效指标问题的取值或编码。
数据审阅说明应为中文。
(四)与监管机构的沟通
申办方根据具体临床试验数据特点及复杂程度,若需要,可按照药物研发与技术审评沟通交流的相关管理办法,与审评机构就临床试验数据库及相关资料的递交进行沟通,以方便审评人员快速、准确地理解申办方递交的临床试验数据。
附录:常用原始数据集
| 数据集 | 命名 | 递交要求 |
|---|---|---|
| 人口学 | dm | 必须递交 |
| 病史 | mh | 如适用 |
| 不良事件 | ae | 如适用 |
| 既往与合并用药 | cm | 如适用 |
| 暴露 | ex | 如适用 |
| 受试者分布 | ds | 如适用 |
| 问卷与量表 | qs | 如适用 |
| 方案偏离 | dv | 如适用 |
| 实验室检查 | lb | 如适用 |
| 心电图 | eg | 如适用 |
| 生命体征 | vs | 如适用 |
| 临床事件 | ce | 如适用 |
| 体格检查 | pe | 如适用 |
附录:研究标签文件
| 标题元素的name属性值 | 说明 |
|---|---|
| data-tabulation-dataset-legacy | 原始数据库(非CDISC标准) |
| data-tabulation-dataset-sdtm | 原始数据库(CDISC标准) |
| data-tabulation-data-definition | 原始数据库数据说明文件、数据审阅说明 |
| analysis-dataset-legacy | 分析数据库(非CDISC标准) |
| analysis-dataset-adam | 分析数据库(CDISC标准) |
| analysis-data-definition | 分析数据库数据说明文件、数据审阅说明 |
| annotated-crf | 注释CRF |
| analysis-program | 编程程序代码 |
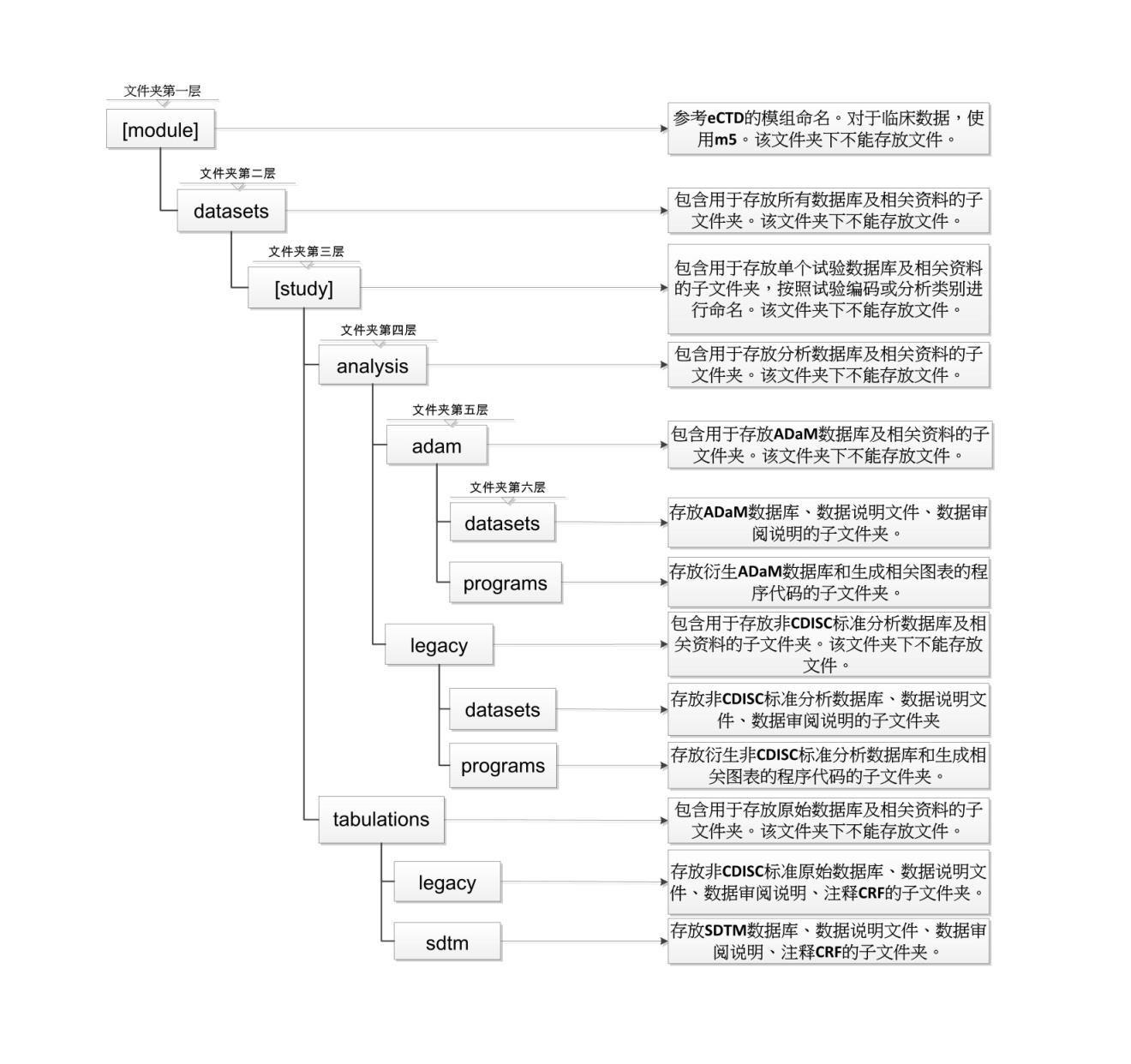
附录:文件夹结构
附录:词汇表
| 术语 | 说明 |
|---|---|
| 编码列表(Code List) | 是指变量可能的取值,包括在试验数据中涉及数据相应的标准编码、行业通用编码或申办方自定义的编码。 |
| 病例报告表(Case Report Form, CRF) | 指按照试验方案要求设计,向申办者报告的记录受试者相关信息的纸质或者电子文件。 |
| 电子通用技术文档(Electronic Common Technical Document, eCTD) | 用于药品注册申报和审评的电子注册文件。通过可扩展标记语言将符合CTD规范的药品申报资料以电子化形式进行组织、传输和呈现。 |
| 数据说明文件(Data Definition File) | 用来描述递交数据的文件,至少应包含递交数据库中各数据集名称、标签、基本结构描述及每一数据集中各变量的名称、标签、类型及来源或衍生过程。 |
| 数据审阅说明(Data Reviewer’s Guide) | 是对数据说明文件的进一步补充,通过文档描述的方式来帮助审评人员更准确、高效的理解与使用所递交的数据库、相关术语、程序代码及数据说明文件信息等。 |
| 注释病例报告表(aCRF) | 是在空白病例报告表的基础上,对采集的受试者数据(电子化的或者纸质的)信息单元(即字段信息)与递交原始数据集中对应的变量或变量值之间映射关系的具体描述。 |
附录:中英文词汇对照
| 中文 | 英文 |
|---|---|
| 便携文档格式 | PDF (Portable Document Format) |
| 病例报告表 | CRF (Case Report Form) |
| 电子通用技术文档 | eCTD (electronic Common Technical Document) |
| 分析数据标准模型 | ADaM (Analysis Data Model) |
| 国际人用药品注册技术要求协调会 | ICH (International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use) |
| 国际万维网协会 | W3C (World Wide Web Consortium) |
| 监管活动医学词典 | MedDRA (Medical Dictionary for Regulatory Activities) |
| 可扩展标记语言 | XML (Extensible Mark-up Language) |
| 可扩展样式表语言 | XSL (Extensible Stylesheet Language) |
| 临床数据交换标准协会 | CDISC (Clinical Data Interchange Standards Consortium) |
| 临床总结报告 | CSR (Clinical Study Report) |
| 受试者水平分析数据集 | ADSL (Subject Level Analysis Dataset) |
| 新药申请 | NDA (New Drug Application) |
| 研究标签文件 | STF (Study Tagging File) |
| 原始数据标准模型 | SDTM (Study Data Tabulation Model) |
| 注释病例报告表 | aCRF (annotated Case Report Form) |
参考文献
[1]. CFDA. 临床试验数据管理工作技术指南. 2016年7月.
[2]. FDA. Study Data Technical Conformance Guide. Mar 2020.
[3]. PMDA. Revision of Technical Conformance Guide on Electronic Study Data Submissions. Jan 2019.
[4]. CDISC. Study Data Tabulation Model Implementation Guide. Nov 2018.
[5]. CDISC. Analysis Data Model Implementation Guide. Oct 2019.